- Visibility 11 Views
- Downloads 3 Downloads
- DOI 10.18231/j.achr.2021.029
-
CrossMark

Hairy cell leukemia - A case report with recent updates
Introduction
HCL and HCL-like disorders like the HCL variant (HCL-V), the splenic marginal zone lymphoma(SMZL), and the splenic diffuse red pulp small B cell lymphoma (SDRPL) are a heterogeneous group of mature lymphoid B-cell disorders which are characterized by the presence of hairy/ villous cells. All these have a specific genetic profile, a different clinical course, and a different treatment strategy. So a correct diagnosis is inevitable. HCL usually presents with a triad of splenomegaly, pancytopenia, monocytopenia, and the characteristic hairy cells on peripheral smear. The BRAF V600E mutation is present in >85% of patients with classic HCL.
Case Report
A 52-year-old male patient was referred to our institution to evaluate pancytopenia, with a history of high-grade fever of 2 weeks duration, not associated with chills and rigor. He had a history of loss of appetite & loss of weight (4kg over one month). He was a known case of Diabetes mellitus detected three years back.
On clinical examination, he was febrile, anemic, and had massive splenomegaly. No peripheral lymphadenopathy was detected, and all other organs were within normal limits
His hemogram showed hemoglobin of 9gm/dl, Total WBC count of 4000/l, Platelet count of 70000 cells/mm3, Liver function test with S.Bilirubin of 1.2mg/dl, and SGOT/SGPT/ALP within normal limits. Cultures, viral serology, and malarial antigen were negative. CT abdomen showed massive splenomegaly (21.7x17.1x9.5 cm) with no hepatomegaly or abdominal lymphadenopathy.
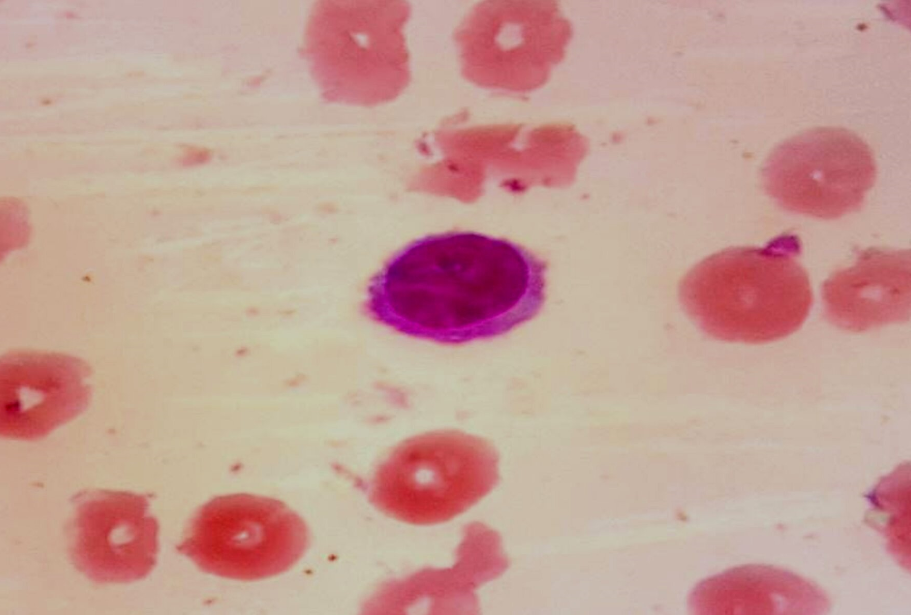
Peripheral smear showed normocytic normochromic anemia, thrombocytopenia, and leucopenia, with 22% of large lymphoid cells having an oval nucleus, homogenous chromatin, inconspicuous nucleoli, moderate amount of pale blue cytoplasm with circumferential hairy projections ([Figure 1])
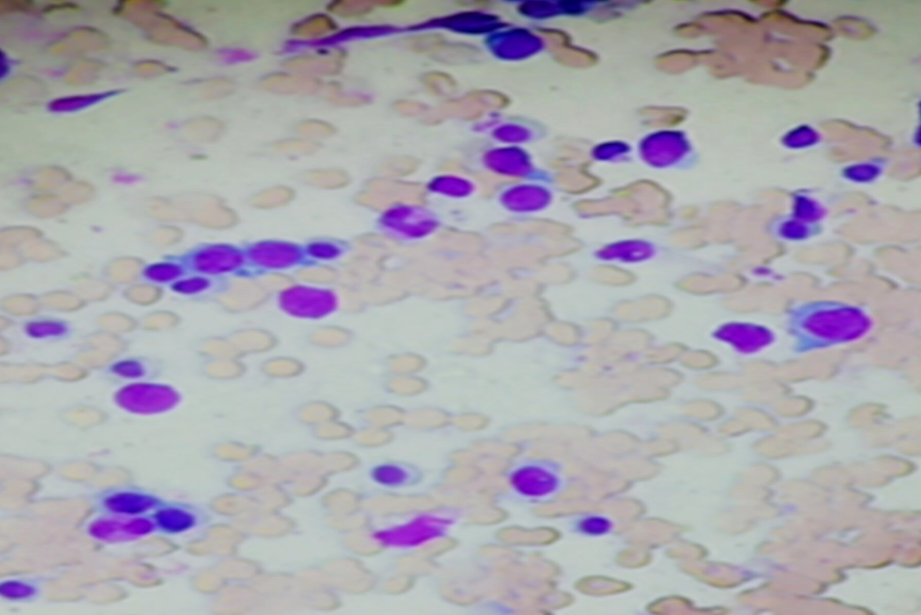
Bone marrow cytology also revealed lymphoid cells ranging in size from 10 to 14 microns with moderate cytoplasm, exhibiting frayed cytoplasmic margins &feathery /hair-like projections along with oval and indented nuclei with inconspicuous nucleoli. These cells were seen interspersed in between normal erythroid and myeloid cells. ([Figure 2]).
Bone marrow biopsy showed hypercellular marrow spaces with monotonous interstitial infiltrate of lymphoid cells having rounded to focally indented nuclei, condensed chromatin, abundant cytoplasm, prominent cell borders producing a fried egg appearance of hairy cells ([Figure 3]). An increase in reticulin fibers was also noted ([Figure 4] )
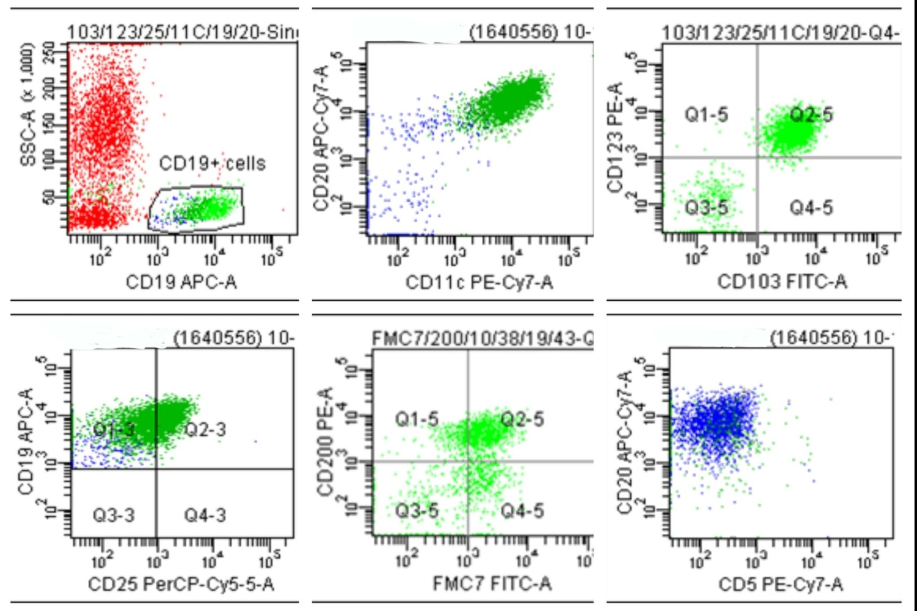
Flow cytometric analysis of CD19 positive singlet cells revealed the expression of bright CD20, with co-expression of CD11c/ CD123/ CD103/ CD25/ FMC7 and negative for CD5.([Figure 5])
With the above clinical, cytomorphological, and immunophenotypic findings, the diagnosis of Hairy Cell Leukemia was made, which was confirmed by BRAFV600E mutation study.
The patient was treated with Cladribine. Minimal residual disease (MRD) analysis and peripheral smear after six months did not show neoplastic cells.
Unfortunately, one year later, the patient developed multi-drug resistant klebsiella and passed away.


Discussion
Hairy cell leukemia is an indolent neoplasm of B cell lymphoid cells with circumferential hairy projections involving peripheral blood and diffusely infiltrating the bone marrow and splenic red pulp. [1], [2] The cell of origin of HCL is possibly the late-activated post-germinal center memory B-cells and the splenic marginal zone B-cells. [3] Usually occur in an age group of 45-65 years with a mean age of 52 years, can range from 20-80 years, 80% of the patients being men. Patients usually present with fever, weight loss, and frequent infections - commonly atypical mycobacterial infections. Lab investigations show pancytopenia (50%)/ monocytopenia. Patients typically have massive splenomegaly, and 55% may have hepatomegaly. The lymph node involvement is usually rare. [2]
Ewald, in 1923 first noticed hairy cells and described them as “Leukamische Reticuloendothelioe.” Later Bouroncle et al.[4] in 1958 represented 26 case series with distinct clinical and pathologic features, and in 1966 Schrek & Donelly used the term “Hairy cell Leukemia” to describe the entity.[5]
Peripheral smear shows pancytopenia and the presence of small to medium-sized hairy lymphoid cells with an oval or indented nucleus, homogenous, spongy, ground glass chromatin, absent/inconspicuous nucleoli, and abundant cytoplasm with circumferential hairy projections.[1], [2] Bone marrow shows diffuse or focal involvement of irregular/ poorly demarcated infiltrates with widely spaced cells.[2], [1] The cells can have a perinuclear halo in formalin-fixed tissue due to abundant cytoplasm. The prominent cell borders give a ‘fried-egg appearance’ with formalin fixation. Mitotic figures are usually rare or absent. A reticulin stain highlights the marked increase in fibrosis, and an increase in mast cells is also commonly seen.[1], [2]
Cytometry with strong granular cytoplasmic positivity for tartrate resistant acid phosphatase(TRAP) is seen. Immunohistochemistry shows positivity for CD20, CD22, CD11C, CD103, CD123, CD25, T bet, Annexin A1, DBA44(CD72), FMC7 and Cyclin D1 and lack CD10 and CD5 expression.[1], [2]
The salient histomorphological and immunophenotypic features of HCL and its differentials are summarized in [Table 1].
Hairy cell morphology is due to the overexpression and activation of members of the RHO family of small GTPases and up-regulation of growth arrest-specific molecule GAS7. Basic fibroblast growth factor (bFGF) and transforming growth factor-beta 1 is the cause for reticulin fibrosis. The minimal lymph node involvement is due to the downregulation of chemokine receptors such as CCR7 and CXCR5.[1], [2]
|
Leukemia / Lymphoma |
Morphology |
IHC |
|
Hairy Cell Leukemia (HCL) |
Hairy cells- Circumferential hairy projections with a homogenous ground glass chromatin, nucleoli are absent/ inconspicuous. Bone marrow infiltrate- patchy / interstitial |
Bright co expression of CD20, CD22 and CD11C with expression of CD103, CD25,CD123, FMC7, Tbet, AnnexinA1, DBA.44 Negative for CD10, CD5 |
|
Hairy cell Leukemia Variant (HCL-V) |
Hairy cells- Circumferential hairy projections with a condensed chromatin and prominent nucleoli. Bone marrow infiltrate- diffuse / interstitial |
Positive for CD20,CD72,CD117,CD103,FMC7 Negative for CD25,CD123, AnnexinA1 |
|
Splenic Marginal Zone Lymphoma (SLVL) |
Villous Lymphocytes- Plasmacytoid with short polar villi. Bone marrow infiltrate – Nodular / interstitial |
Positive for CD20, CD79a Negative for CD23, CD103, CD43,Cyclin D1, CD5, CD10, AnnexinA1 |
|
Splenic Diffuse Red Pulp Small B Cell Lymphoma (SDRPL) |
Villous lymphocytes- plasmacytoid with short polar villi, vesicular chromatin, and occasional distinct nucleoli. Bone marrow infiltrate- intrasinusoidal / nodular / interstitial |
Positive for CD20, CD72 Negative forCD25, CD123,CD23,CD11c |
The various genomic alterations:[3]
MAPK Pathway - BRAF V600E somatic mutation was found in a patient with HCL by using whole-exome sequencing (WES) in 2011 and constitute 70- 100% of the cases. The other rare alterations include MAP2K1: Cell cycle- COKN1B (p27): NFKB pathway – KLF2 : Spliceosome- TP53: NOTCH Pathway - NOTCH 1:
Epigenetic regulators- KMT2C (histone methyltransferase), ARID1A (SWI/SNF family), KDM6A (histone demethylase), CBEBBP : TF repressor - BCOR.
90% of HCL patients have a mutated immunoglobin gene heavy chain variable rearrangement (IGHV) profile.[3]
Treatment strategies
Treatment is needed if the patients are symptomatic or if the hematologic parameters are declining. The hematologic parameters indicating a need for treatment include at least one of the following: hemoglobin <11 g/dL; platelet count <100 G/L; absolute neutrophil count <1 G/L. Symptomatic splenomegaly may serve as an indication for treatment.[5]
Chemotherapy with Purine nucleoside analogs (PNA) like Cladribine (2-CdA) or Pentostatin (DCF) is the standard first-line treatment for HCL, conferring in most cases a more prolonged overall survival. The second-line therapy is by Cladribine/ Pentostatin with Rituximab and Bendamustine.[4], [5]
The uncontrolled infections require Alpha-Interferon (IFN-α), which is also used as an alternative in pregnant women.
Relapsed/ Refractory cases with BRAF V600E mutations are treated with BRAF inhibitors like Vemurafenib, Dabrafenib, or Moxetumomab/ Pasudotox or BRAF inhibitors plus MEK inhibitors like Trametinib/ Cometinib. In BRAF wild-type relapsed/ refractory cases, Moxetumomab/ Pasudotox is used.[4], [5].
Splenectomy may be considered in patients with resistant massive symptomatic splenomegaly with a low-level bone marrow infiltration and pregnant patients with progressive HCL and those refractory to nucleoside analogs and IFN-α. [6], [7]
Allogeneic stem cell transplantation is recommended in younger patients with multiple relapses and refractory to purine analogs and Rituximab.[8]
The response criteria are summarized in [Table 2].
|
Response Categories |
Criteria |
|
Complete Response |
Absence of hairy cells on peripheral blood, bone marrow aspiration or biopsy specimens, and the normalization of organomegaly and peripheral blood count. |
|
Partial Response |
The normalization of peripheral blood counts, at least50% reduction in organomegaly and bone marrow hairy cells, and <5% circulating hairy cells. |
|
Relapse |
Any deterioration in blood counts related to the detection of hairy cells in peripheral blood and/or bone marrow and/or increasing splenomegaly |
The minimal residual disease (MRD) can be done after 4-6 months of therapy.
Flow cytometry, using an 8 color panel (CD103/CD305/CD19/CD123/CD25/CD3/CD45/CD20), can be a useful and sensitive tool for detecting blood MRD. [1]
Prognosis
Massive splenomegaly, leukocytosis with an increased number of hairy cells in the peripheral blood, and an immunoglobulin heavy chain variable region gene mutation are considered the worst prognosis. The VH4-34 positive cases are often associated with a poor prognosis.[9]
As the patients are at increased risk of developing second cancers like Hodgkin Lymphoma, Non-Hodgkin lymphomas, and thyroid cancer, a close follow-up is highly recommended. The increased risk may be related to the disease and/or the treatment. The role of Pentostatin or Cladribine in the development of secondary malignancies remains debatable.[9]
Source of Funding
No financial support was received for the work within this manuscript.
Conflict of Interest
The authors declare they have no conflict of interest.
References
- T. Robak, E. Matutes, D. Catovsky, P.L. Zinzani, C. Buske. Hairy cell leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015. [Google Scholar] [Crossref]
- S Swerdlow, E Campo, N L Harris. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: International agency for research on cancer. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: International agency for research on cancer 2017. [Google Scholar]
- SH Swerdlow, E Campo, NL Harris, ES Jaffe. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th Edn. 2008. [Google Scholar]
- X Troussard, E Cornet. Hairy cell leukemia 2018: Update on diagnosis, risk-stratification, and treatment. Am J Hematol 1002. [Google Scholar]
- BA Bouroncle, BK Wiseman, CA Doan. Leukemic Reticuloendotheliosis. Blood 1958. [Google Scholar] [Crossref]
- LA Andritsos, MR Grever. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015. [Google Scholar] [Crossref]
- J Jansen, J Hermans, . Splenectomy in hairy cell leukemia: A retrospective multicenter analysis. Cancer 1981. [Google Scholar] [Crossref]
- BA Adeniji, M Fallas, M Incerpi,, S Hamburg, R Katz, D Ogunyemi. Laparoscopic splenectomy for hairy cell leukemia in pregnancy. Case Rep Med 2010. [Google Scholar] [Crossref]
- L Zp, F B Al, P C. Hairy cell leukemia: allogeneic transplantation could be an optimal option in selected patients. Clin Lymphoma Myeloma Leuk 2012. [Google Scholar]